Blog
Introducing the next generation of InstaNovo models

Announcing InstaNovo v1.1
Since our InstaNovo paper is now published, we’d like to share an update on what we’ve been working on while our manuscript was under review. With the release of our preprint over a year ago, we were overwhelmed by the response from the community and the applications of our models. We are actively collaborating with experts and continue to explore the potential of solutions for de novo peptide sequencing. Our ongoing efforts focus on developing more accurate models, expanding the de novo sequencing ecosystem for analysis and data reporting, fine-tuning our models, and designing tailored, application-specific workflows.
While much of this work is still in progress, we have just released an improved version of our base model, InstaNovo v1.1. This model boasts higher recall, greater identification certainty, expanded support for modifications, and enhanced data processing and reporting features. We believe these advancements are worth communicating with this post instead of an article, and we are excited to show you how this model compares to the earlier model in our paper.
Getting started
Our new model is available in the main InstaNovo branch with detailed documentation on installation, local execution, and running it in training or testing mode with your data. If you prefer a hosted solution, you can access the model via our HuggingFace space, where you can upload your files and analyse them on free GPU compute. To reproduce the analysis in this blog post or to run this analysis on your own dataset, refer to this Jupyter notebook.
Training data
InstaNovo v1.1 (IN v1.1) has been trained on the ProteomeTools, MassiveKB, and kind dataset contributions from several other projects processed by the CompOmics group at Ghent University. We are grateful for the huge community effort to generate, process, and curate the datasets that have enabled the development of our models. We are excited to contribute with the release of this combined dataset in the near future.
Benchmarking IN v1.1
IN v1.1 is a substantial improvement over IN v0.1, the original model from our paper. To benchmark these models, we used a standard HeLa proteome run, a widely used reference sample in proteomics facilities for quality control and one of the validation datasets in our study.
Compared to the gold standard database search, IN v0.1 achieved a recall of 49.5%. IN v1.1, using greedy precursor mass fit search (i.e. selecting the sequence that best fits the observed precursor mass), improves recall to 56.7%, while knapsack beam search further boosts recall to 63%. This compounds to a 13.5% improvement over the previous model, with more than six out of ten peptides being detected without precursor mass filtering on predictions (Figure 1a). Additionally, IN v1.1 predicts 42.6% more Peptide Spectrum Matches (PSMs) that map to the proteome with exact sequence matching (Figure 1b).
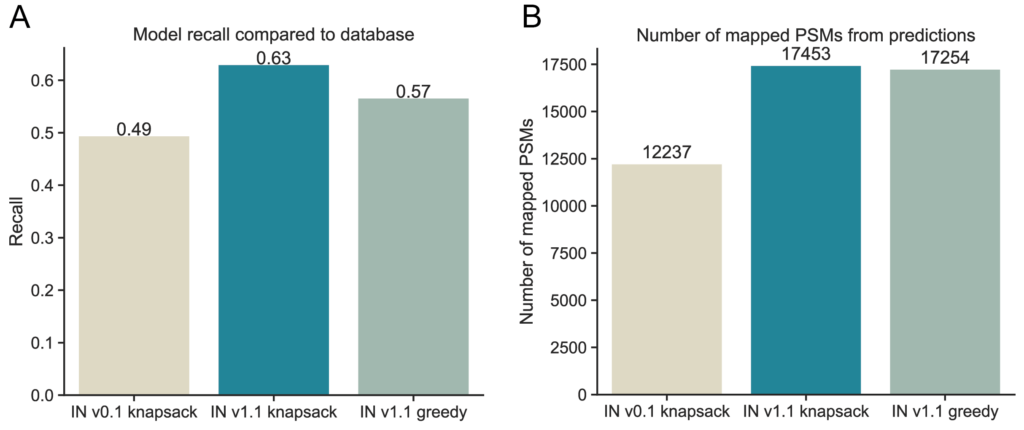
We use peptide-level metrics and especially recall to assess our models, the most direct evaluation of prediction performance. This is because bottom up proteomics is a peptide centric methodology, where the percent accuracy within the peptide chain is not particularly informative. Crucially, we evaluate whether the complete sequence prediction is correct.
The distribution of model confidence, defined as the product of our residue log probabilities raised in its natural exponent, indicates that the new model is sharper in its prediction certainty. We observe more high confidence predictions being correct, while lower confidence predictions are more densely clustered in the lower confidence range (Figure 2a). Accordingly, we observe better precision values in parallel with increased coverage (Figure 2b).

While it is still unclear whether this trend will persist with larger training datasets and increasing model sizes, this pattern suggests that confidence-based thresholding can help reduce false positive rates.
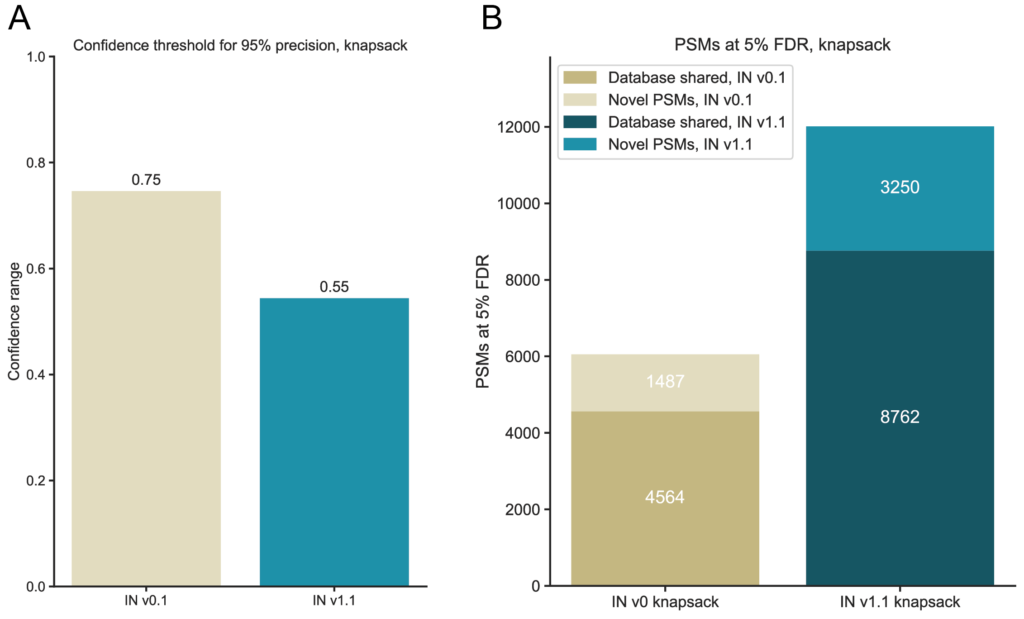
We use this precision to estimate the false discovery rate (FDR) and derive confidence cutoffs for low false identifications, which yield identification results aligned with conventional proteomics search outputs. In agreement with our confidence analysis, we observe that lower confidence thresholds are required to maintain a 5% FDR (Figure 3a). At the same threshold, IN v1.1 identifies more novel PSMs compared to its predecessor (Figure 3b).
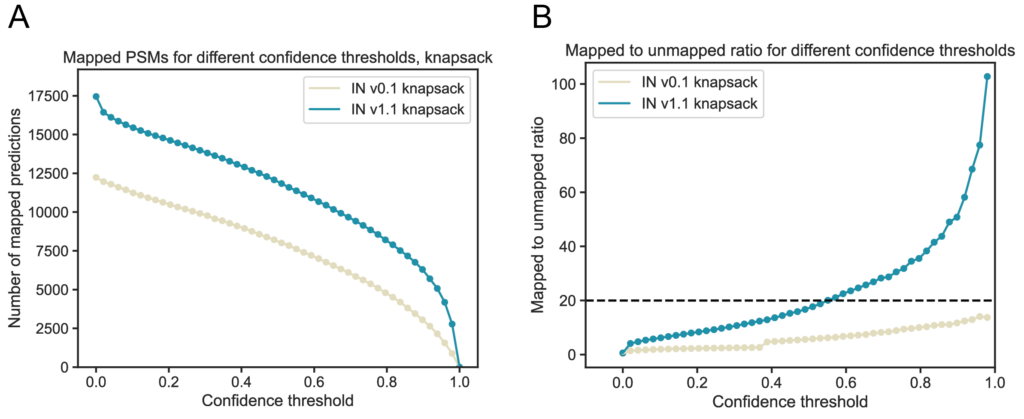
To assess FDR more directly, we mapped predicted PSMs to the human proteome. As expected, we observed an increased proportion of PSMs mapping to the proteome, with the greatest improvements in the high confidence range (Figure 4a). Additionally, when comparing the ratio of mapped to unmapped predictions, IN v1.1 demonstrates a considerable reduction in false positive rates (Figure 4b, dashed line indicates 5% FDR when directly mapping to proteome without database grounding).
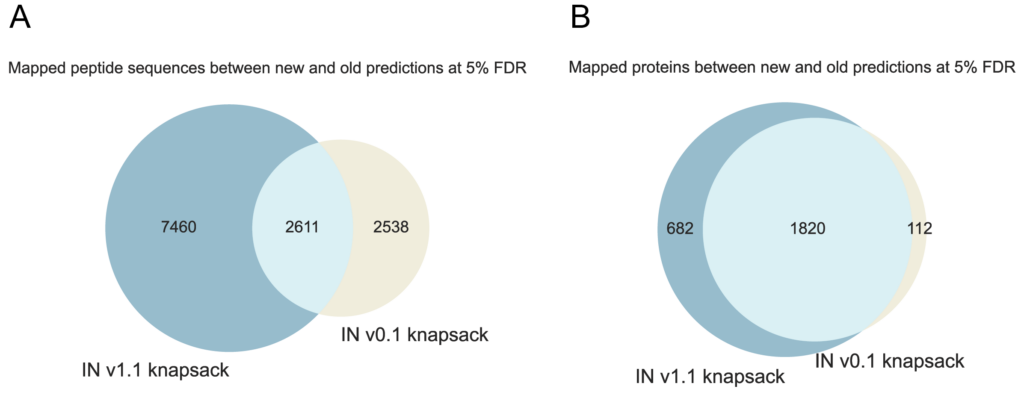
Due to this higher discrimination of false positives and PSMs in the model confidence scale, IN v1.1 identifies 145.1% more peptides mapping to the proteome at 5% FDR compared to its precursor (Figure 5a), leading to a 35.3% increase in protein identifications with the same threshold (Figure 5b).
Expanded modification support, features and runtime
The IN v1.1 release introduces compatibility with four additional modifications: phosphorylation, deamidation, carbamylation, and ammonia loss. Carbamidomethylation is now a variable modification, with variable acetylation and oxidation also supported. This results in an increase in overall peptide sequence predictions (Figure 6a), with a substantial number of high confidence predictions at 5% FDR, even in samples without enrichment (Figure 6b). For large datasets, especially when prioritising high-confidence results, there is a tradeoff between performance and inference time (Figure 6c). Despite linear scaling, knapsack precursor fit takes 36 times longer than greedy precursor fit per spectrum. While knapsack beam search provides the best performance, it comes at a steep computational cost. In many cases, running the model with greedy precursor mass search may be more efficient while still delivering robust results.

In the updated version, a spectrum dataframe class has been introduced to further improve data import robustness and interoperability. Residue log probabilities are now included in the results alongside the token list, and scan number and precursor mass error (in ppm) are now also included in the output table. The model also supports evaluation mode with a database search reference, where FDR confidence thresholds are automatically reported during inference. In the new model, leucine and isoleucine are predicted as separate residue tokens, albeit with relatively low recall at peptide level (17.7% for leucine and 19.6% for isoleucine).
Performance on other datasets and outlook
Although here we focus on a single dataset, we have observed similar performance gains across biological samples and applications (Figure 7), indicating generalized gains. Importantly, we observe a 81.3% peptide recall in our GluC dataset, which is a GluC pretreated HeLa proteome with more data than the HeLa QC above.
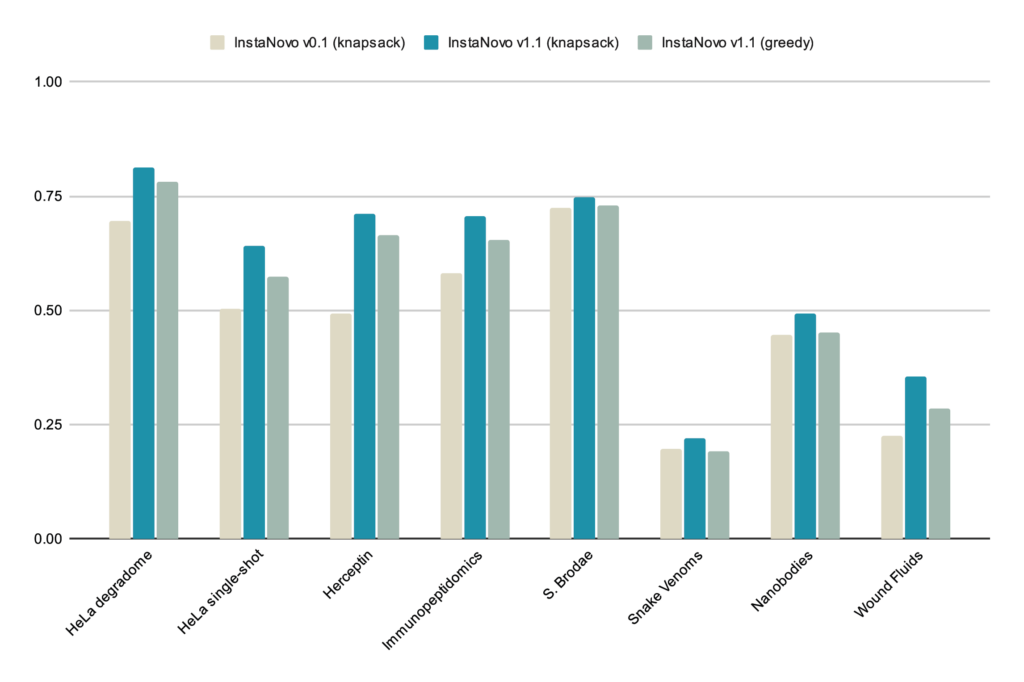
We believe our models continue to improve in performance, and we have yet to determine the upper limit for database search recall and novel identification rates. Our goal is to achieve robust accuracy near the 90% recall mark across experimental datasets, edging closer to solving de novo peptide sequencing. We will continue to scale and refine our models, as well as explore biological applications and features further. Additionally, the next generation of IN+, our diffusion model, is being trained and will be released soon.
So stay tuned for more updates, several preprints coming out soon!
Source of all images: internal
Original research paper available at: InstaNovo enables diffusion-powered de novo peptide sequencing in large-scale proteomics experiments